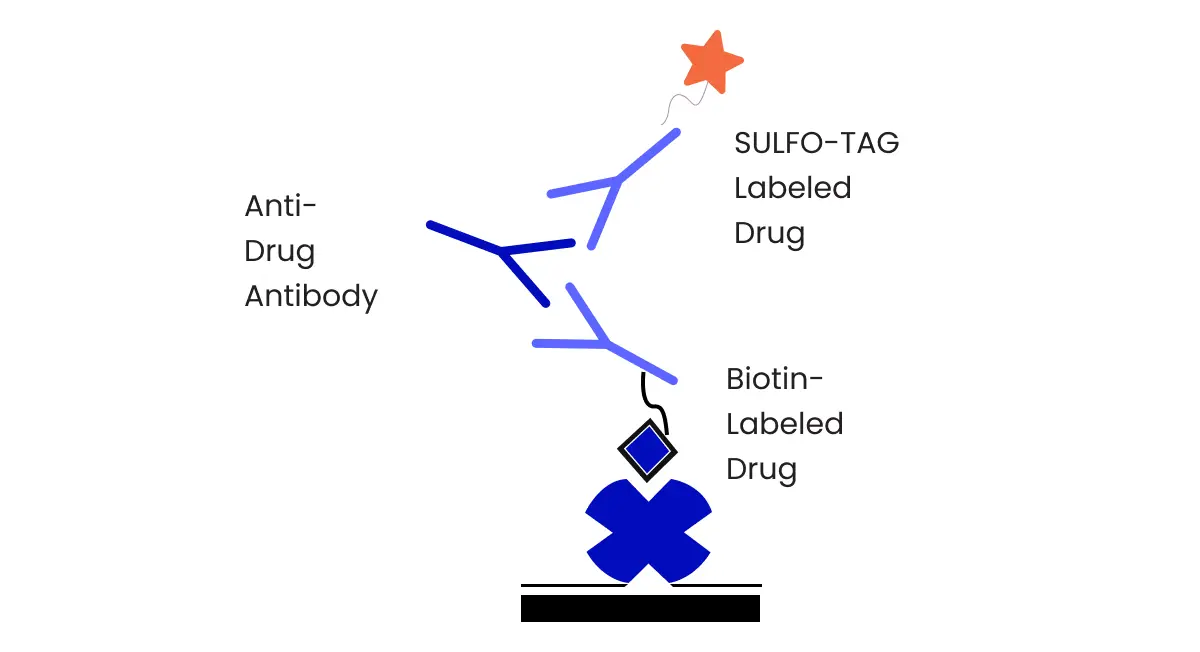
Overview & Definition: Drug Development Pathway
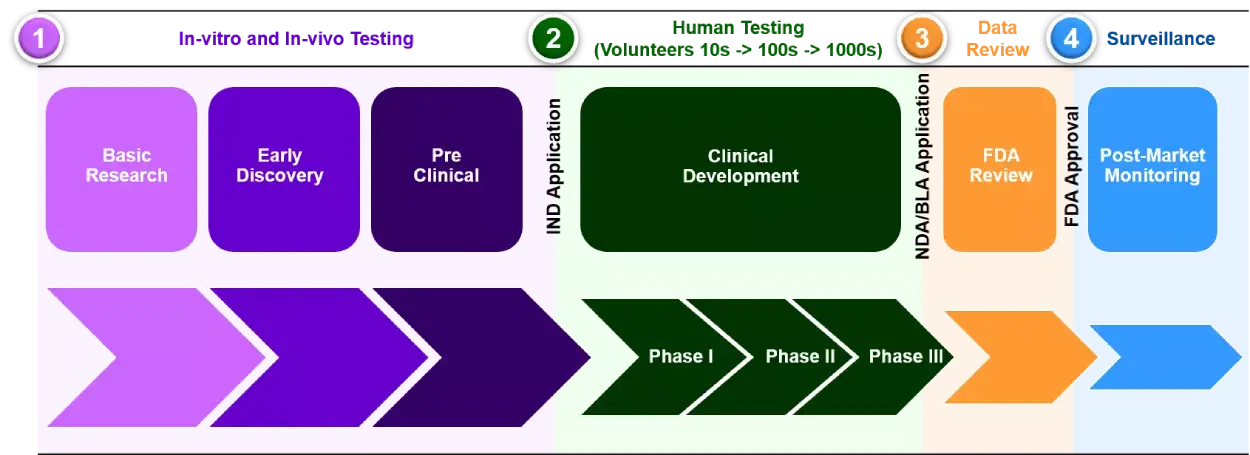
The complexity in drug development has increased manifolds over the past 40 years, requiring preclinical phase of drug development, investigational new drug (IND) application, and complete clinical testing before marketing approval from the FDA. Generally, new drug applications (NDAs) or biologics license applications (BLA) are reviewed comprehensively before approval, and then drug performance is resubmitted to regulatory agencies for post-marketing studies. The overarching goal is to bring more efficient and safer treatments to the patients as quickly as possible after a thorough medical evaluation.
Phases And Stages
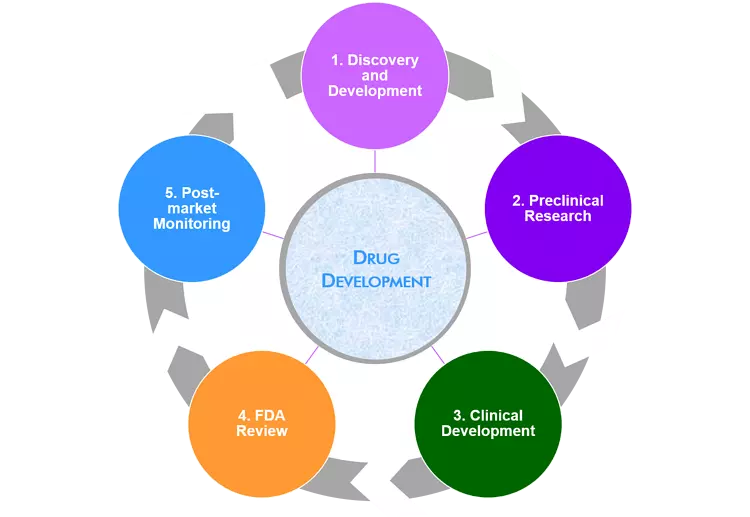
There are five critical steps in the U.S. FDA drug development process, including many phases and stages within each of them. We will discuss each phase and different stages of drug development to develop an in-depth understanding of the entire process. The phases of drug development are –
- Step 1: Discovery and Development
- Step 2: Preclinical Research
- Step 3: Clinical Development
- Step 4: FDA Review
- Step 5: FDA Post-Market Safety Monitoring.
Step 1: Discovery & Development

Drug discovery research is how new medications are discovered. Historically, drug discovery, design, and development mostly started with identifying active ingredients from traditional medicines or purely by chance. Later, classical pharmacology was used to investigate chemical libraries including small molecules, natural products, or plant extracts, and find those with therapeutic effects. Since human DNA was sequenced, reverse pharmacology has found remedies to existing diseases through modern testing.
Disease processes, molecular compound tests, existing treatments with unanticipated effects, and new technologies spur drug discovery timeline through the cycle below.
Today the steps in drug discovery and development involve screening hits, iterative medicinal chemistry, and optimization of hits to reduce potential drug side effects (increasing affinity and selectivity). Efficacy or potency, metabolic stability (half-life), and oral bioavailability are also improved in these steps of the drug development process.
Target Identification And Validation
Target identification finds a gene or protein (therapeutic agent) that plays a significant role in disease. Afterward, scientists and researchers record the target’s therapeutic characteristics. Drug targets must be efficacious, safe, usable, and capable of meeting clinical and commercial requirements. To validate targets, researchers use modern tools and techniques such as disease association, bioactive molecules, cell-based models, protein interactions, signaling pathways analysis, functional analysis of genes, in vitro genetic manipulation, antibodies, and chemical genomics. For example, the Sanger Whole Genome CRISPER library and Duolink PLA are excellent sources for drug discovery targets.
Hit Discovery Process
Following target validation, compound screening assays are developed.
Assay Development And Screening
Assay development in drug discovery is a crucial component of drug discovery workflow. Assays are test systems that evaluate the effects of the new drug candidate at the cellular, molecular, and biochemical levels.
High Throughput Screening
High Throughput Screening (HTS) uses robotics, data processing/control software, liquid handling devices, and sensitive detectors to rapidly conduct millions of pharmacological, chemical, and genetic tests, eliminating hours of painstaking testing by scientists. HTS identifies active compounds, genes, or antibodies that affect human molecules.
Hit to Lead
In the Hit to Lead (H2L) process, small molecule hits from an HTS are evaluated and optimized in a limited way into lead compounds. These compounds then move on to the lead optimization process.
Lead Optimization
In the lead optimization (LO) process, the lead compounds discovered in the H2L process are synthesized and modified to improve potency and reduce side effects. Lead optimization conducts experimental testing using animal efficacy models and ADMET tools, designing the drug candidate.
Active Pharmaceutical Ingredients
Active pharmaceutical ingredients (APIs) are biologically active ingredients in a drug candidate that produce desired effects. All drugs are made up of the API or APIs and excipients. Excipients are inactive substances that deliver the drug into the human system. High Potency Active Pharmaceutical Ingredients (HP APIs) are molecules that are effective at much smaller dosage levels than standard APIs. They are classified based on toxicity, pharmacological potency, and occupational exposure limits (OELs), and used in complex drug development involving more than ten steps.
The drug discovery process gets narrowed when one lead compound is found for a drug candidate, and the process of drug development starts.
Step 2: Preclinical Research

Once a lead compound is found, preclinical phase of drug development begins with in vivo research to determine the efficacy and safety of the drug. Researchers determine the following about the drug:
- Absorption, distribution, metabolization, and excretion information
- Potential benefits and mechanisms of action
- Best dosage, and administration route
- Side effects/adverse events
- Effects on gender, race, or ethnicity groups
- Interaction with other treatments
- Effectiveness compared to similar drugs
Preclinical trials test the new drug on non-human subjects for efficacy, toxicity, and pharmacokinetic (PK) information. These trials are conducted by scientists in vitro and in vivo with unrestricted dosages.
Absorption, Distribution, Disposition, Metabolism, & Excretion
Absorption, Distribution, Disposition, Metabolism, & Excretion (ADME) is a Pharmacokinetic (PK) process of measuring the ways the new drug affects the body. ADME involves mathematical descriptions of each effect.
Proof of Principle / Proof of Concept
Proof of Principle (PoP) are studies that are successful in preclinical trials and early safety testing. Proof of Concept (PoC) terminology is used almost interchangeably with PoP in drug discovery and development projects. Successful PoP/PoC studies lead to program advancement to the Phase II studies of dosages.
In Vivo, In Vitro, And Ex Vivo Assays
These three types of studies are conducted on the whole, living organisms or cells, including animals and humans; or using non-living organisms or tissue extract. In vivo, preclinical research examples are the development of new drugs using mice, rat, and dog models. In vitro is research conducted in a laboratory. Ex vivo uses animal cells or tissues from a non-living animal. Examples of ex vivo research assays are finding effective cancer treatment agents; measurements of tissue properties (physical, thermal, electrical, and optical); and realistic modeling for new surgical procedures. In an ex vivo assay, a cell is always used as the basis for small explant cultures that provide a dynamic, controlled, and sterile environment.
In Silico Assays
In silico assays are test systems or biological experiments performed on a computer or via computer simulation. These are expected to become increasingly popular with the ongoing improvements in computational power, and behavioral understanding of molecular dynamics and cell biology.
Drug Delivery
New drug delivery methods include oral, topical, membrane, intravenous, and inhalation. Drug delivery systems are used for targeted delivery or controlled release of new drugs. Physiological barriers in animal or human bodies may prevent drugs from reaching the targeted area or releasing when they should. The goal is to prevent the drug from interacting with healthy tissues while still being effective.
- Oral: Oral delivery of medications is reliable, cost-effective, and convenient for patients. Oral drug delivery may not monitor precise dosages to the desired area but is ideal for prophylactic vaccinations and nutritional regimens. Delayed action, stomach enzyme destruction, absorption inconsistencies, or patients with gastrointestinal issues or upset can occur, and patients must be conscious during administration.
- Topical: Topical drug delivery involves ointments, creams, lotions, or transdermal patches that deliver a drug by absorption into the body. Topical delivery is more useful for patient skin or muscular conditions — it is preferred by patients due to non-invasive delivery and their ability to self-administer the medicine.
- Parenteral (IM, SC or LP Membrane): Parenteral drug delivery utilizes bodily membranes, including intramuscular (IM), intraperitoneal (IP), or subcutaneous or (SC). It is often used for unconscious patients and avoids epithelial barriers that are difficult for drugs to cross.
- Parenteral (Intravenous): Intravenous injection is one of the fastest drug delivery absorption methods. IV injection ensures entire doses of drugs enter the bloodstream, and it is more effective than IM, SC, or LP membrane methods.
- Parenteral (Inhalation): Inhalation drug delivery gets the drug rapidly absorbed into the mucosal lungs, nasal passages, throat, or mouth. Problems with inhalation delivery include difficulty delivering the optimum dosage due to small mucosal surface areas and patient discomfort. Pulmonary inhalation drug delivery uses fine drug powders or macromolecular drug solutions. Lung fluids resemble blood, so they can absorb small particles easily and deliver them into the bloodstream.
Formulation Optimization & Improving Bioavailability
Formulation optimization is ongoing throughout pre-clinical and clinical stages. It ensures drugs are delivered to the proper place at the right time and in the right concentration. Optimization may include overcoming solub
Step 3: Clinical Drug Development Process
Once preclinical research is complete, researchers move on to clinical drug development, including clinical trials and volunteer studies to finetune the drug for human use.
Complexity of Study Design, Associated Cost & Implementation Issues
The complexity of clinical trial design and its associated costs and implementation issues may affect trials carried out during this phase. Trials must be safe and efficacious and be completed under the drug development budget, using a methodology to ensure the drug works as well as possible for its intended purpose. This rigorous process must be set up correctly and enroll many volunteers to be effective.
Clinical Trials – Dose Escalation, Single Ascending & Multiple Dose Studies
Proper dosing determines medication effectiveness, and clinical trial examine dose escalation, single ascending, and multiple dose studies to determine the best patient dosage.
Phase I – Healthy Volunteer Study
This phase is the first time the drug is tested on humans; less than 100 volunteers will help researchers assess the safety and pharmacokinetics, absorption, metabolic, and elimination effects on the body, as well as any side effects for safe dosage ranges.
Phase II and Phase III – Studies in Patient Population
Phase II assesses drug safety and efficacy in an additional 100-500 patients, who may receive a placebo or standard drug previously used as treatment. Analysis of optimal dose strength helps create schedules while adverse events and risks are recorded. Phase III enrolls 1,000-5,000 patients, enabling medication labeling and instructions for proper drug use. Phase III trials require extensive collaboration, organization, and Independent Ethics Committee (IEC) or Institutional Review Board (IRB) coordination and regulation in anticipation of full-scale production following drug approval.
Biological Samples Collection, Storage & Shipment
During clinical trials, biological samples are collected, stored, and shipped from testing sites according to global standards and regulations. Transport containers of biological samples may include dry ice packs or other temperature stabilizing methods. Different requirements apply to different types of biological samples.
Pharmacodynamic (PD) Biomarkers
PD biomarkers are molecular indicators of the drug’s effects on the target human area, and link drug regimen and biological responses. This data can help select rational combinations of targeted agents and optimize drug regimens and schedules. Rationality and hypothesis-testing power are increased through the use of PD endpoints in human trials.
Pharmacokinetic Analysis
Pharmacokinetic analysis is an experimental trial that determines the theory of how a new drug behaves in the human body. The volume of distribution, clearance, and terminal half-life are defined through compartmental modeling.
Bioanalytical Method Development and Validation
Bioanalytical methods detect analytes and metabolites such as drug or biomarkers in biological or human samples to determine drug efficacy and safety. The complete bioanalytical assay consists of sample collection, clean-up, analysis, and detection.
Drug (Analyte) & Metabolite Stability in Biological Samples
Stability is important in determining human drug efficacy, and biological samples are required. Drug and drug metabolites are susceptible to degradation, which can lower drug concentration over the life of the drug.
Blood, Plasma, Urine & Feces Sample Analysis for Drug and Metabolites
Biological samples used in clinical trials include blood, plasma, urine, and feces to determine and analyze various properties and effects of the drug and its metabolites on humans.
Patient Protection – GCP, HIPAA, & Adverse Event Reporting
Human patients must always be protected during clinical trials, and Good Clinical Practices (GCP), the Health Insurance Portability and Accountability Act (HIPAA), and adverse event reporting to IEC/IRB regulates and ensures their safety.
Step 4: FDA Review

Once the new drug has been formulated for its best efficacy and safety, and the results from clinical trials are available, it’s advanced forward for wholistic FDA review. At this time, the FDA reviews and approves, or does not approve, the drug application submitted by the drug development company.
Regulatory Approval Timeline
The new drug regulatory approval timeline may be standard, fast track, breakthrough, accelerated approval, or priority review depending on its applications and necessity for patients. If standard or priority review is required, the approval timeline may be up to an year. Fast track, breakthrough, or accelerated approvals may occur sooner.
IND Application
IND applications are submitted to the FDA before starting clinical trials. If clinical trials are ready to be conducted, and the FDA has not responded negatively about the drug, developers may start the trials.
NDA / ANDA / BLA Applications
An NDA abbreviated new drug application (ANDA), or BLA is submitted to the FDA after clinical trials demonstrate drug safety and efficacy. The FDA reviews study data and decides whether to grant approval or not. Additional research or an expert advisory panel may be required before a final decision is made.
Orphan Drug
An orphan drug is intended to treat disease so rare that financial sponsors are unwilling to develop it under standard marketing conditions. These drugs may not be approved quickly or at all.
Accelerated Approval
New drugs may be granted accelerated approval if there is strong evidence of positive impact on a surrogate endpoint instead of evidence of impact on actual clinical benefits the drug provides. Expedition of approval means the medication can help treat severe or life-threatening conditions.
Reasons for Drug Failure
New drug applications may fail for a variety of reasons, including toxicity, efficacy, PH properties, bioavailability, or inadequate drug performance.
- Toxicity: If the toxicity of a new drug is too high in human or animal patients, the drug may be rejected due to safety concerns about its use following manufacture.
- Efficacy: If a new drug’s efficacy is not high enough or evidence is inconclusive, the FDA may reject it.
- PK Properties or Bioavailability: PK properties or poor bioavailability due to low aqueous solubility, or high first-pass metabolism, may also cause a drug to fail FDA review. PK causes of drug failure include inadequate action duration and unanticipated human drug interactions.
- Inadequate Drug Performance: If the new drug performs the desired function, but only at a shallow level, the FDA may reject the application in favor of a formulation that performs better.
Step 5: Post-market Monitoring
Following drug approval and manufacturing, the FDA requires drug companies to monitor the safety of its drug using the FDA Adverse Event Reporting System (FAERS) database. FAERS helps FDA implement its post-marketing safety surveillance program. Through this program, manufacturers, health professionals, and consumers report problems with approved drugs.
Here’s a summary of the FDA drug approval process discussed thus far.
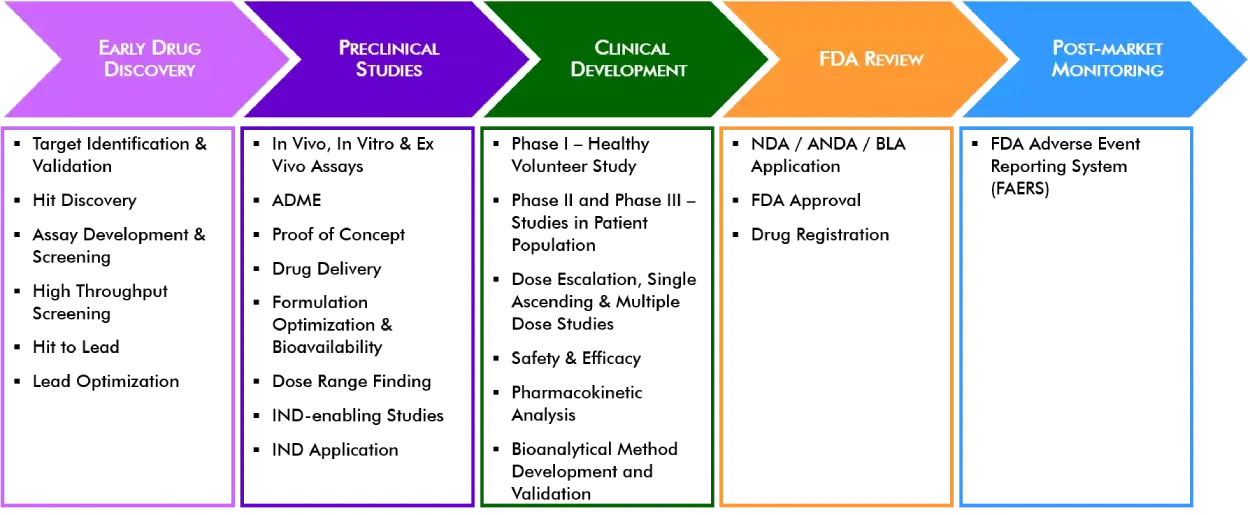
Other Relevant Drug Development Concepts
Drug Master File
A Drug Master File (DMF) is a submission to the FDA used to provide confidential, detailed information about facilities, processes, or articles used in the manufacturing, processing, packaging, and storing of a human drug.
Drugs for Pediatric Use
Drugs for pediatric use are intended for use in children or youth, generally under the age of 21. In some cases, the American Academy of Pediatrics (AMA) may make exceptions if a pediatrician and family agree on an older age adult.
Drugs for Veterinary Use
Drugs for veterinary use are intended for use in animals, pets, and livestock. However, some veterinary drugs get their start in humans and then change to human and animal drugs.
Small Molecule vs. Biologics
Small molecules have a variety of applications or biological functions. Large molecules (also called biologics) are proteins with a therapeutic effect. Small molecules work by cell signaling. Large molecule drugs are complex and may be composed of over 1,300 amino acids. They are identical versions of human proteins.
Process Scale-up Differences & Difficulties
Drug development involves generating progressively larger medicine batch sizes, and changes in processes for different-sized batches may cause unexpected difficulties. Use of the right pharmaceutical equipment can be helpful, as well as the discovery of parameters that affect critical process parameters (CPPs).
New drug development is a highly regulated, complicated process that requires specialists and intense research and development skill sets in the medical research community. All regulations and safety indications must be observed carefully, and human and animal clinical trials subjects treated professionally and with the utmost care. The goal of drug development is to prevent human and animal pain and suffering whenever possible and find and provide new drugs that we can depend on to improve our health and happiness.
Conclusion
Lastly, what do Sheryl Crow, Lance Armstrong, and Robert De Niro have in common? They are all among the lucky ones who survived the deadly disease cancer. There’ll be thousands in the current year alone who wouldn’t be as fortunate, though. An estimated 1.74M+ new cancer cases will be diagnosed, and 600k+ cancer deaths would be reported. All of us in the life sciences sector can’t rest on our laurels and must continue innovating faster to find a cure for this nemesis of humanity.