Overview & Definition
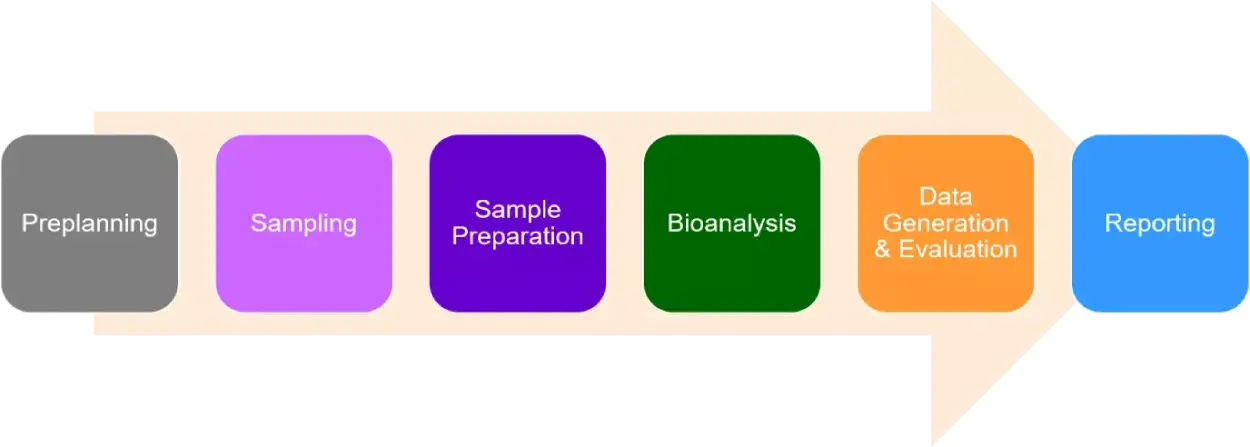
Bioanalysis isn’t merely running standard analyses for a biological sample. We must produce results which are quantitative and valid as per FDA guidance as these analyses constitute the foundational block towards drug approval. Due to the significance of these analyses, regulatory authorities generally audit these results for accuracy before approval of the drug.
It’s imperative to have the support and expertise of veterans, such as the team at NorthEast BioLab, when the complexities of bioanalysis get cumbersome. Our scientists help navigate bioanalytical method validation following method development focused on the appropriate detection range for your assay, as well as offer insights on the requirements of FDA and other international regulatory authorities. This article alludes how our team can help you with custom assay development that meets all the criteria outlined above.
Sample Preparation
We often go through several steps to clean up and prepare the sample before it is ready for LC-MS/MS analysis, in a process typically referred to as sample extraction. These initial sample preparation steps help remove any molecules or substances that interfere with the analyte detection, ultimately leading to significant improvements in analyte detection, selection and instrument performance.
Sample Characterization
Target molecules for bioanalysis, or analytes in chemistry verbiage, fall into one of two broad categories – small molecules or large molecules. Small molecules are typically synthesized or isolated from natural sources, have lower molecular weights, and are usually assayed using chromatographic assays chemical entities (drugs), metabolites, or pharmacodynamic. Large molecules include higher molecular weight peptides, proteins, nucleic acids, lipids, and polysaccharides, and are usually assayed using ligand binding assays.
While there are a plethora of different instrumental techniques that can be used for bioanalysis, we primarily use the highly sensitive and sophisticated liquid chromatography instrument coupled to tandem mass spectrometry (LC-MS/MS) and ELISA for small and large molecules, respectively, among other technologies. Our expertise in handling biological fluids extends to serum, plasma, urine, cerebrospinal fluid (CSF), organ tissues, etc. Our team can effectively quantify your drugs, and their metabolites as well as pharmacodynamic biomarkers from complex biological matrices.
Protein Removal
Proteins and phospholipids are one of the most common interferents found in biological samples while measuring biomarkers, drugs, and metabolites. Proteins can precipitate and clog a chromatographic column if left unremoved from these samples. Proteins can also bind to the small molecules of interest, which may include the analyte, preventing an accurate measurement of analyte concentration. Commonly used techniques for protein removal include protein precipitation (PPT), liquid-liquid extraction (LLE), and solid phase extraction (SPE).
Phospholipid Removal
We know that there are numerous phospholipids in biological samples such as plasma – a common type of biological sample for drug metabolism and pharmacokinetic (DMPK) studies. Phospholipids are organic molecules that are found in cell membranes and are composed of a hydrophilic head group which contains phosphate and choline, plus a hydrophobic fatty acid chain tail. Phospholipids interfere with the reliability of data collected by LC-MS/MS with electrospray ionization (ESI) because, as surfactants, phospholipids aggregate at the surface of the droplets formed during the liquid-to-gas transition of ions. With the phospholipids covering the surface of the droplets, the analyte ions are less able to escape the droplet which suppresses analyte detection. Therefore, to improve analyte detection, the removal of phospholipids before analysis is often essential. Commonly used methods for phospholipid removal from samples include LLE and SPE, although some phospholipids can be removed from a sample during PPT as well.
Protein Precipitation
PPT removes proteins from a sample by denaturing the proteins causing them to precipitate. Proteins can be denatured through the addition of organic solvent, an acid, a base, or heat to a sample. Acetonitrile and methanol are the most commonly used organic solvents for PPT. Once the protein has precipitated, it is pelleted by centrifugation, and the supernatant is used for analysis. One downside of this technique is that samples are diluted, and sensitivity may be compromised. To precipitate as much protein as possible, we add at minimum two or three equivalents of organic solvent for each equivalent of the sample. PPT can also be followed by solvent evaporation and reconstitution of the analyte in an appropriate solution or buffer and offset dilution of sample.
Liquid-liquid Extraction
LLE is a sample preparation technique that is widely used in the analysis of drugs. LLE begins with the addition of an immiscible organic solvent to a biological sample that’s typically aqueous. The analyte is extracted from proteins and lipids into the organic solvent layer while the proteins and lipids remain in the aqueous phase. Neutrally-charged analytes tend to be the most effectively extracted using LLE, but the addition of an inorganic salt to a mixture of a biological sample and a water-miscible organic solvent causes the two solutions to separate and allows hydrophilic analytes to partition into the organic solvent phase. While LLE is quite useful for salt removal from samples, it has some limitations regarding the number of analytes that can be simultaneously extracted based on dispersion between the polarities of each analyte.
Solid Phase Extraction
SPE uses solid sorbent material packed into a column or disc to efficiently and selectively remove interferents from biological samples. When a sample is applied to the packed sorbent material, both analytes and interferents will adsorb to the material. Interferents are washed off the sorbent material using solvents such as acetonitrile or methanol. When acetonitrile is used as a solvent, phospholipids tend to be more readily removed from the sample, whereas the use of methanol tends to lead to the presence of phospholipids in the sample. After interferents are washed from the sorbent material, the analyte(s) are eluted and collected. Typically, either reverse phase or ion exchange SPE cartridges are used in drug bioanalysis.
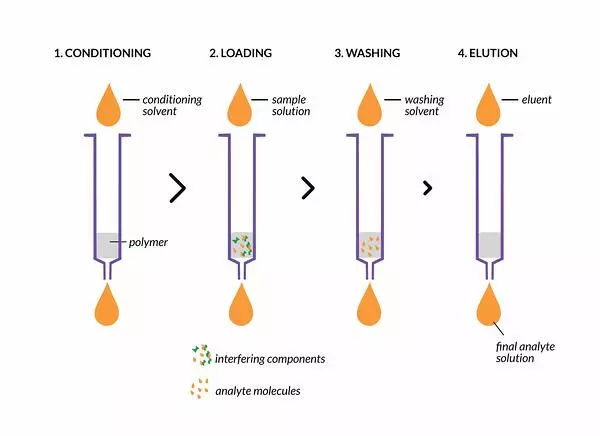
On-line Extraction and Direct Injection
Method sensitivity is increased given lower sample dilution when sample preparation or extraction techniques such as SPE are connected on-line with the analysis instrument. Reproducibility improves as well since the analyte no longer needs to be collected and injected into the instrument.
Sample preparation can be time-consuming and can produce large amounts of waste chemical material, so it can be tempting to inject samples into the instrument for analysis directly. However, a lack of sample cleanup tends to lead to contaminants and physical clogging in the apparatus, so often on-line extraction will be added to the instrument to provide sample cleanup before analysis.
Other Endogenous Material Removal
Other endogenous molecules that are commonly found in biological samples include polysaccharides, salts, nucleic acids, metabolites in urine, and secondary metabolites. Because these molecules can also modify the signal response of the analyte of interest, they are removed via methods such as filtration, centrifugation, or simple dilution. Metabolites are very important as they may have the same daughter ion in MS and can possibly interfere with analyte detection. Metabolites can be removed during sample cleanup step, but in most cases, it is preferred to collect metabolites in the sample as it is often important to quantitate metabolites as well. Ideally, robust bioanalytical method development and validation aims to separate and reliably detect a drug and its metabolite(s) in a single method.
DBS testing
Dried blood spot (DBS) testing involves the collection of a blood sample by pricking either a heel or a finger and blotting blood onto high-quality filter paper. The blood can be eluted from the paper using buffer to dissolve the dried blood before analysis, or the blood can be analyzed directly from the paper using desorption electrospray ionization (DESI) or direct analysis in real time (DART).
Bioanalytical Method Development
Robust bioanalytical method development and validation is key to accurate measurements of drugs, metabolites, and biomarkers in bioanalytical assays. Several underlying steps are adequately completed during bioanalytical research. Above all, we must determine and consider the nature and any relevant characteristics of the sample itself. For example, the composition of the sample and the expected concentration range of the analyte constitute a couple of noteworthy criteria among several others to be considered. The FDA, EMA, ICH, and other regulatory agencies provide bioanalytical method development guidelines that ensure adherence to the principles required for reliable bioanalytical methods.
Step-by-step Protocol
First, we define the goal or purpose as this usually drives the method design. Next, we determine the analyte concentration range and select the reference standard to set the boundaries of the method design space. Consequently, we get ready to create the detailed method steps while following established standard operating procedures (SOPs). Figuring out the experimental matrix and sampling plan help to fill in procedural details for testing and analyzing critical study parameters. At this point, the method can be tested and optimized for bioanalytical validation and actual sample analysis.
Selectivity
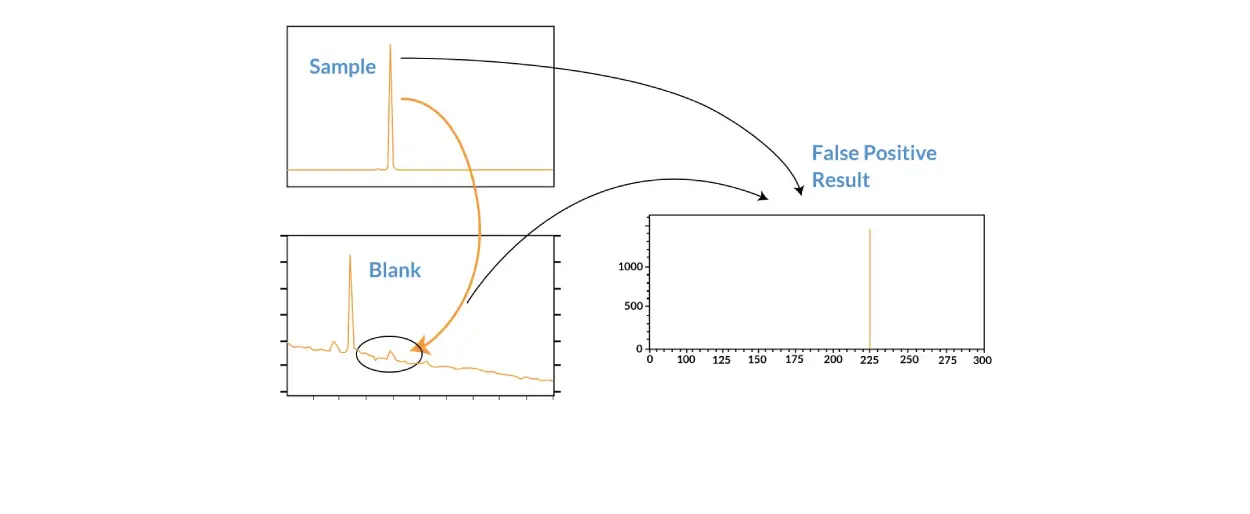
The selectivity of a method is its ability to differentiate between the analyte, the internal standard, and any other biological matter found in the sample. The selectivity of the method is determined by analyzing at least six blank samples from differing sources. Blank samples comprise of the biological matrix without any analyte or internal standard.
Carry-over Issues
Carry-over is the presence of the analyte in a blank sample analysis that has been conducted following a high-concentration sample, and needs to be addressed during the bioanalytical development for any chromatographic assay Analyzing a blank sample after a high-concentration sample or after the upper limit of quantification (ULOQ) calibration standard will show if carry-over is an issue for your analysis or not. Carry-over in a blank sample should not exceed 20% of the lower limit of quantification (LLOQ).
Lower Limit of Quantification (LLOQ)
The lower limit of quantification (LLOQ) is the lowest concentration of the calibration standards that is not zero and provides a measure of sensitivity. The LLOQ is the lowest concentration of the analyte that can be quantified reliably. The signal generated by the analyte at the LLOQ should be greater than or equal to five times the signal generated by the zero-calibration standard.
Calibration Curve
For chromatographic assays calibration curve is prepared by measuring the response from a set of prepared calibration standards that increase linearly in concentration. Calibrators are prepared similarly for ligand binding assays, but the response may be non-linear and require more complex mathematical fitting. Calibration standards are prepared by spiking blank matrix solution with a known concentration of reference standard. The calibration curve should have at least six concentrations of calibration standards plus a blank (a matrix that has been through the sample preparation process but does not contain either an analyte or internal standard) and a zero (blank containing internal standard). Each calibration standard should meet pre-defined criteria for accuracy, precision, and selectivity. The calibration curves can be monitored over repeated batch assays to ensure bioanalytical assays are accurate, precise, and reproducible.
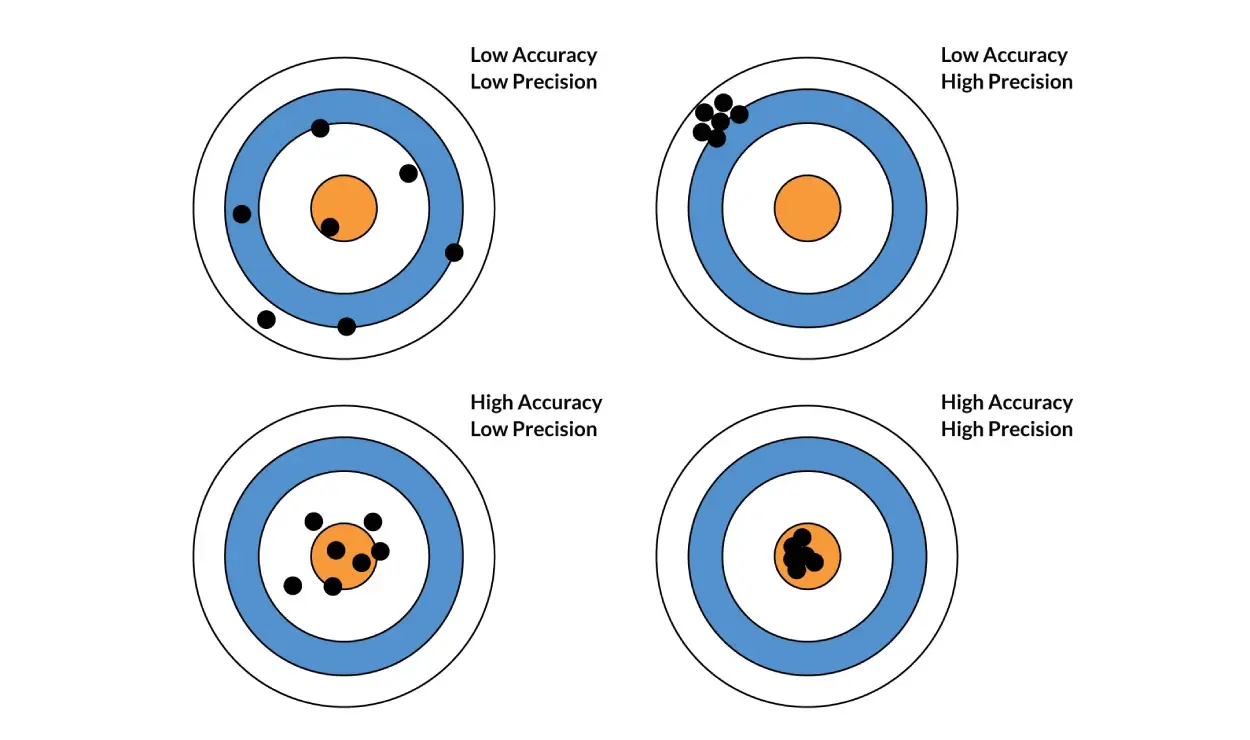
Accuracy vs. Precision
Accuracy is defined by the closeness of the measured analyte concentration to the actual concentration of the analyte in the sample. Precision is the closeness of repeated measurements of analyte concentration to each other. In bioanalytical research, both accuracy and precision are measured using Quality Control samples (QC). Accuracy is expressed as a percentage and is acceptable if at least five QC standard replicates at three concentration levels are within 15% of the nominal concentrations for chromatographic assays and 20% for ligand binding assays during bioanalytical method development and validation. Precision is expressed as the coefficient of variation (CV; also known as relative standard deviation, RSD) and is acceptable if the CV is within 15% for chromatographic assays and 20% for ligand binding assays.
Dilution Integrity
If a sample needs to be diluted to fit within the detection range of the instrument, it’s vital to ensure that the act of diluting the sample will not affect the accuracy and precision of the measurement during bioanalytical analysis. To test whether dilution affects accuracy and/or precision (the dilution integrity), a sample of blank matrix should be spiked with analyte at a concentration above the upper limit of quantification (ULOQ) and then diluted with blank matrix to within the range of the study dilutions and then tested for accuracy and precision values Dilution integrity is tested during bioanalytical validation and appropriate controls are included in all sample runs where dilutions are included.
Reference Standards
For any bioanalysis, a well-characterized solution of an analyte, known as a reference standard, is used to prepare calibration standards and quality control (QC) standards in the biological matrix. Calibration standards are used to generate the calibration curve, and quality control samples are used to measure the performance of the method. Calibration and QC standards can be prepared either by diluting reference standard in organic solvent and then spiking that into the biological matrix or by spiking the reference standard into biological matrix and further diluting the standards with more matrix.
The reference standard should be obtained from a reliable source and should be received with a certificate of analysis (CoA) that demonstrates identity and purity, as well as information such as the batch/lot number, storage conditions, and expiration or retest date.
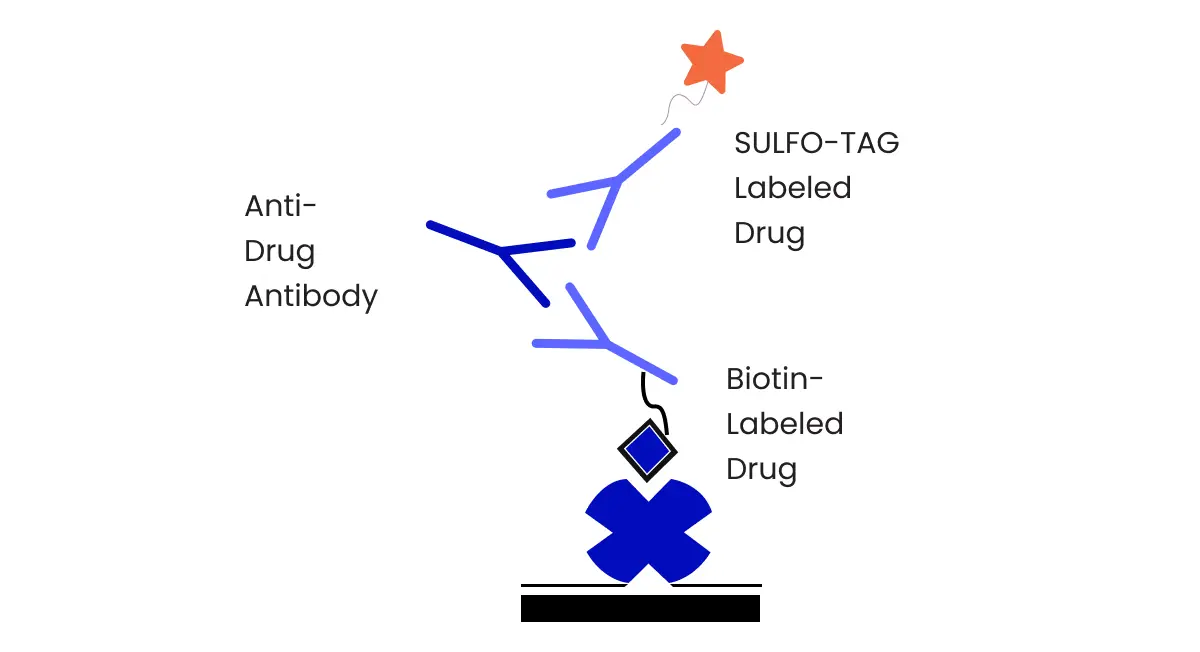
For chromatographic assays, a structurally similar analog of the analyte is generally used as Internal Standard, but a stable labeled isotope analyte of the molecule is an ideal internal standard if available when using mass spectrometry detection. Most importantly an ideal internal standard must not interfere with the analyte and a stable labeled isotope internal standard must not contain over 1-2% of unlabeled analyte as that can interfere with quantitation.
Matrix Effects
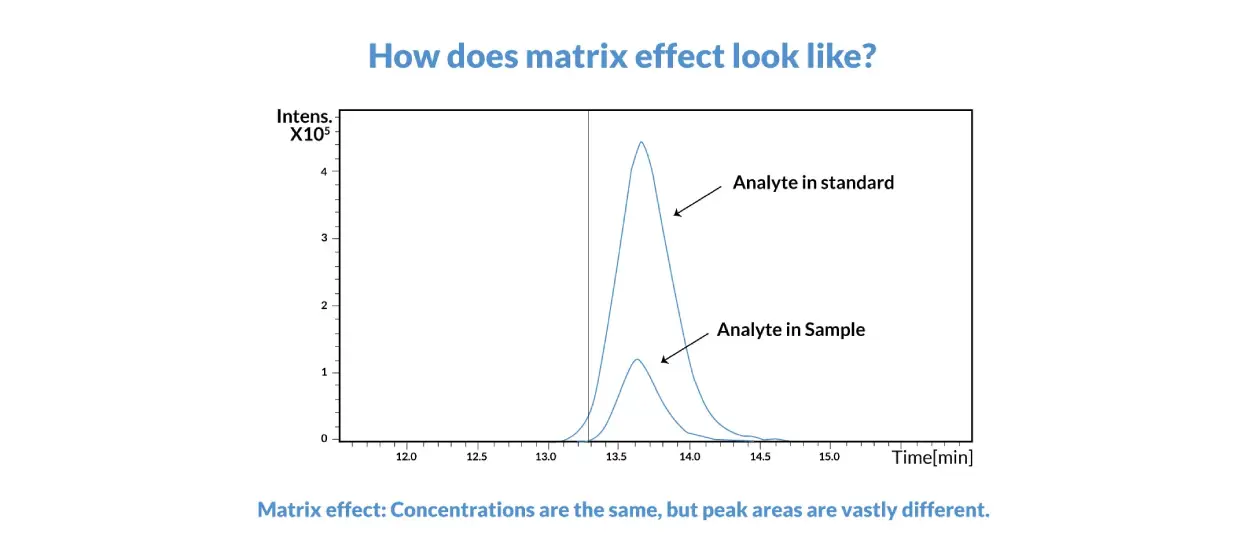
Matrix effects occur when any molecule (for example, metabolites or decomposition products, vehicle excipients, or endogenous xenobiotics) other than the analyte that is found in the sample changes the way the analyte responds during detection. It’s possible for matrix effects to cause either matrix suppression or enhancement of the analyte signal. A value called the matrix factor (MF) can be calculated by determining the peak area of a sample of the blank matrix that has undergone sample preparation and then been spiked with an analyte and determining the peak area of a sample of a pure analyte. Simply put, the ratio of these two values gives the MF. To normalize the MF to the internal standard, divide the MF calculated for the analyte by the MF calculated for the internal standard. The impact of matrix effects is minimized during sample preparation for bioanalytical assays since calibrators and QCs are typically prepared in blank matrix matching that of the test samples. Understanding the matrix effect is useful during bioanalytical method development in order to target the appropriate calibration range and detection limits for sample analysis.
Stability Testing
Analyte stability is determined to ensure that sample handling and storage don’t affect the analyte concentration and therefore the analysis results in a specific sample matrix. Quality control samples are analyzed under varying sample handling conditions including storage at room temperature, in autosampler, during freeze-thaw cycle, and over long-term storage at specific temperatures vs. when freshly prepared, and the results are compared. The values determined should be within 15% of the nominal concentration to demonstrate adequate stability under the various conditions.
At NorthEast BioLab, we commonly test stability at room temperature for bench-top stability, as well as at -20 °C and at -70 °C for freeze-thaw and long-term storage stability.
Bioanalytical Method Validation

Full Validation
The FDA, ICH, and other regulatory agencies provide bioanalytical method development and validation guidelines outlining the required parameters for appropriate bioanalytical development, validation, and analysis. A full validation study should be performed for any custom assay method that is new, or that has been developed based on the literature. A fully validated method shows that it can be used to determine the concentrations of analytes such as a drug and its metabolite(s) reliably in a specific biological sample matrix from a particular species. The key characteristics that need to be determined for the performance of a method to be considered acceptable are sensitivity selectivity, reproducibility, intra- and inter-day accuracy and precision, dilutional integrity, matrix effects, and analyte stability under all storage and sample handling and preparation conditions.
Partial Validation
There are times when a full method validation need not be performed. For example, when a method is based on a previously validated method where only minor changes have been made. Partial validation must be run when an analysis is performed in another laboratory or using different equipment, or when the sample origin or storage conditions have changed. A partial validation may be as straightforward as determining the accuracy and precision of the modified method.
Cross-Validation
A cross-validation study must be performed if data has been put together using different methods in a study or if data has been collected in different laboratories. Essentially, cross-validation study allows the data to be reliably compared across disparate sources.
Sample Analysis

Aliquoting
Aliquoting is the portioning of a sample into precisely known amounts (such as volume or mass) to be used in further analyses. Knowledge of the exact quantity of the aliquot is important for any quantifications made during sample analysis. It is vital to maintain a standard procedure for aliquoting samples to ensure consistency and accuracy.
Analytical Run
An analytical run is the analysis of a single batch of samples. A batch of samples is a complete set of samples along with calibration standards, QC samples, and matrix blanks that have been processed at the same time in the order in which they will be analyzed. The analysis run-time varies widely based on the methodology and instrumentation as an analytical run may take 24+ hours to complete, or multiple analytical runs may be completed within a single day.
Acceptance Criteria of an Analytical Run
The acceptance criteria of an analytical run is defined in the method development SOP. For chromatographic assays, one standard criterion is a calibration standard accuracy level where at least 75% of calibration standards should be within 15% of the nominal concentration of the standard determined by back-calculation (using the calibration curve to find out the concentration of the calibration standards). Another standard criterion is a QC standard accuracy level where at least 67% of QC standards should be within 15% of their nominal concentrations for chromatographic bioanalytical assays.
Calibration Range
The calibration range is determined early in method development and may be re-evaluated later during sample analysis due to a narrower range of analyte concentrations in samples than expected, or due to analyte concentrations of samples being outside of the calibration range. If the calibration curve range is changed, the method should be partially revalidated to ensure accuracy and precision.
Reanalysis of Study Samples
Samples may be reanalyzed for any number of reasons, including the rejection of the analytical run or malfunction of the equipment. Reasons for the reanalysis of study samples should be outlined in the SOP during method development, and the number of samples that were reanalyzed should be included in the study report.
Incurred Sample Reanalysis (ISR)
An incurred sample is one taken from a subject that has been dosed, and incurred sample reanalysis, a requirement of the bioanalytical method validation guidance for industry, supports the reliability and reproducibility of the reported data as the accuracy and precision of real samples adds to the data collected from QC standards.
Integration
The parameters used for signal integration should be outlined in the SOP and should be maintained in laboratory documentation. Any deviations should be discussed in the bioanalytical report.
Reporting

Records must be produced and securely stored to ensure proper method validation. Based on the validation and bioanalytical reports, a study should be able to be repeated as reported.
Validation Report
The bioanalytical method validation ppt and report should contain all the necessary information concerning the validation that was performed. The SOPs created during method development should be referred to in the validation report for any needed method details. All the source data should be available upon request in the original format. Any deviations from the validation protocol should be reported.
Bioanalytical Report
Reference should be made to the validation report in the bioanalytical method development ppt and report, and otherwise, the bioanalytical method development report deals with details of the method and samples, as well as the actual data generated.
Regulatory Guidelines

With our team of dedicated scientists, we develop and optimize bioanalysis methods on which we then perform assay validation as per FDA and EMA guidelines using Good Laboratory Practices (GLP), regulated under US 21CFR part 58 and the bioanalytical method validation guidance for industry.
FDA and EMA Guidance on Method Validation
Both the Food and Drug Administration (FDA) and the European Medicines Agency (EMA), provide bioanalytical method validation guidelines based on GLP principles. For clinical studies bioanalytical method validation ICH guidelines are followed. By following these regulatory guidelines, a fully validated bioanalytical method is guaranteed to produce data that is accurate and reliable.
Assay development performed at NorthEast BioLab is always validated following the most recently published FDA (2018) and EMA (2011) guidelines.
Good Laboratory Practices (GLP)
Good Laboratory Practice (GLP) is a regulatory concept that was introduced in the 1970s in the USA due to concerns over the validity of data provided in new drug applications. As a quality control system, GLP principles offer a tool to ensure that laboratory studies are planned, conducted, and documented to a minimum and rigorous standard for data quality and validity. GLP principles also define the responsibilities of facilities and personnel operating in a GLP-compliant bioanalytical lab.
The Organization for Economic Co-operation and Development (OECD) has harmonized GLP principles for international use to allow for the acceptance of previously generated data during the trade of chemicals.
Conclusion
As you can see, bioanalytical testing is a complicated endeavor that provides incredibly valuable information about the safety and efficacy of drugs in a trustworthy manner.
Common applications of bioanalysis performed at NorthEast BioLab include testing drug and metabolite exposures, bioavailability, bioequivalence, pharmacokinetics, and toxicokinetics, and diagnostic biomarkers in various studies conducted during preclinical (animal) and clinical (human volunteers) phases of drug development. Similarly, Immunogenicity tests are also considered bioanalytical methods as their targeted analytes are antibodies in serum or plasma.
For expert support in developing and validating bioanalytical methods for your drug development and research, contact us at NorthEast BioLab about our bioanalytical laboratory services. We seek contentment in serving our clients towards their noble mission of bettering the current standards of treatments.