Overview & Definition
Closely related fields in the biochemical pharmacology discipline include clinical pharmacokinetics, pharmacodymamics, pharmacology, and toxicokinetics. Clinical Pharmacokinetics is the clinical application of basic PK principles, involving the design and modification of dosing regimens in line with therapeutic drug pharmacokinetics monitoring. Pharmacodynamics meaning defines the relationship between plasma and tissue drug and/or metabolite concentrations, time, and therapeutic response.Understanding the pharmacokinetics vs pharmacodynamics definition is crucial for integrating these concepts into effective therapeutic strategies.
Simply put, PK describes what the body does to the drug, and pharmacodynamics meaning describes what the drug does to the body. Pharmacology studies help us understand the influence of the drug on the body.Pharmacology studies help us understand the influence of the drug on the body, which ties into the broader pharmacokinetics vs pharmacodynamics definition. In the context of pharmacodynamics vs pharmacokinetics, pharmacodynamics focuses on the drug’s effects on the body, whereas pharmacokinetics addresses how the body affects the drug. Toxicokinetics involves the relationship of the principles of drug PK to the disposition of toxicants and their metabolites and the time course of toxic or adverse events in the body. When considering pharmacodynamics vs pharmacokinetics, it’s important to note that pharmacokinetics evaluations are used throughout the drug discovery and development processes to aid in the understanding of dosing requirements, levels necessary for the desired therapy, and potentials towards toxicity, adverse effects, and drug-drug or drug-food interactions. Understanding pharmacodynamics vs pharmacokinetics is crucial in distinguishing how drugs behave in the body versus how the body responds to drugs, particularly in the context of toxicokinetics, and this understanding is central to the pharmacokinetics vs pharmacodynamics definition.
PK in Early Drug Discovery
Early drug pharmacokinetics discovery processes typically involve the synthesis and testing of numerous test articles with the goals of achieving disease target interaction with specificity, selectivity, and potency. PK drug testing is used in preclinical efficacy species (typically mice) during early drug pharmacokinetics discovery to determine whether a potential drug has the necessary exposure to achieve efficacy following in vivo PK dosing. These early drug PK studies are used to demonstrate sensible exposures for interpretation of target proof-of-concept – is the test article target specific, does it have adequate systemic exposure to reach the therapeutic target to achieve efficacy, and does the interpreted biochemical mechanism of action make sense relative to the observed exposure and efficacy in early pharmacokinetic and preclinical pharmacology studies.
Related in vitro ADME assessments are often also useful in a compound screening cascade. Prior to in vivo PK dosing, in vitro experiments may be performed to predict metabolic stability, cell permeability potential, protein binding, and drug-drug interaction potentials. Understanding pharmacokinetics vs pharmacodynamics ADME is crucial for determining how a compound’s absorption, distribution, metabolism, and excretion relate to its therapeutic effects and safety profile. These experiments may be used to filter out compounds with undesirable metabolic liabilities, poor absorption or lack of ability to reach the therapeutic target or high potential for metabolic interactions with other drugs or xenobiotics in the body.
PK in Late Drug Discovery
Near the end of lead compound optimization and identification, a select number of preclinical candidate molecules are further characterized in preparation for preclinical toxicology and development. Synthetic chemistry processes may change for batch scale-up, and improvements or changes in compound purity and crystallinity may affect PK exposures in animals. PK with scaled up batches in these cases should be tested for any differences from prior batches, and additional species are often tested to better understand single dose drug PK in the efficacy model species and planned species for preclinical toxicology and toxicokinetics assessments. PK may be compared using differing formulations and may be investigated for additional interactions and effects, for example fed vs. fasted states or evaluations of gender differences. Drug accumulation and steady-state assessments are often made by comparing PK following multiple doses to a single dose. When considering pharmacokinetics vs pharmacodynamics ADME, it is important to understand how the drug’s absorption, distribution, metabolism, and excretion influence its therapeutic effects and safety profile. Higher doses may be tested to establish values for maximum tolerated dose and determination of therapeutic index, calculated as the ratio of maximum tolerated dose to minimum efficacious dose. If different animal strains are used for efficacy testing, they may be compared as well. PK data may be evaluated in tandem with efficacy model endpoints to better understand compound PD and PK relationships. Pharmacokinetics vs pharmacodynamics ADME studies in late discovery support the planning for toxicology as well, with predictions of potential metabolic liabilities, differences in cross-species metabolism, and potentials for drug-drug interactions.
Preclinical Toxicology and Toxicokinetics
The US FDA and other national regulatory agencies require a minimum of two species for preclinical toxicology studies, including one rodent (rat, guinea pig) and one non-rodent (dog, monkey, minipig). In the vast majority of cases, toxicology studies include assessments of compound toxicokinetics as well as PK PD in drug development in order to aid in the interpretation of any observed toxicological effects. Doses for toxicology are typically higher than those used in earlier PK assessments and efficacy studies, ranging from the minimum efficacious dose to the maximum tolerated dose. All aspects of toxicological and toxicokinetic assessments in these studies are regulated and conducted following 21 CFR part 58 (Good Laboratory Practice for Nonclinical Laboratory Studies).
Clinical Pharmacokinetics
While various techniques such as allometry may be used to predict human PK based on scaling data from preclinical species results, pharmacokinetics meaning in clinical trial studies are ultimately required to relate doses to systemic exposure and efficacy, which is central to understanding the pharmacokinetics meaning in a clinical context. In early clinical trials, sub-efficacious doses are initially tested in healthy volunteers to ensure safety and get initial PK parameter estimates, and scaled up as appropriate to efficacious levels. In these cases, PK may be evaluated at each cohort with medical monitoring prior to dosing higher levels to ensure systemic exposures remain below any limitations that may have been discovered during preclinical toxicology. This step is crucial to ensuring there are no adverse events that would require limiting or suspending further dosing in a patient or volunteer, thereby emphasizing the pharmacokinetics meaning in terms of balancing safety and efficacy during drug development. In select cases, such as a chemotherapeutic agent for an aggressive cancer treatment, initial trials may be in a disease population, since the drug pharmacokinetics could risk healthy volunteers, is intended to treat a life-threatening disease, and may be beneficial to patients who haven’t responded to other treatments. Such cases are regulated by the FDA and other global regulatory agencies. For generics or reformulations of previously approved and marketed drugs, bioequivalence studies are required in order to ensure that the drug exposure of the new formulation or product yields the same exposures at the same dose levels as the approved product. Bioequivalence is assessed by comparing specific parameters such as pharmacokinetics Cmax, AUC, and bioavailability of the two products following dosing.
What is Pharmacodynamics?
Pharmacodynamics is a division of biochemical pharmacology that defines the relationship between plasma and tissue drug and metabolite concentrations, time, and biological response. The goal of pharmacodynamics studies is to understand what the drug does to the body in terms of therapeutic response and toxicity, which is essentially the pharmacodynamics meaning in clinical terms. Pharmacodynamic studies are often evaluated in tandem with PK analysis to better understand the relationship of exposure and effect and the timing of therapeutic or toxicological responses. PK assays in target tissues are often performed to better understand the pharmacodynamics meaning in terms of serum or plasma to better understand the relationship of systemic and target tissue exposures and the relationship between exposure and response, referred to as the PD and PK relationship.
Pharmacokinetics Study Designs
The key components to any pharmacokinetic study are the test model (species, strain, gender, age, healthy or disease state), number of subjects, test compound, route of dosing administration, dosing regimens, dosing vehicle, sample matrix, and sample collection time course. Other factors to consider in a pharmacokinetic study may include controls to ensure compound stability in the collected samples prior to analysis, anesthesia used prior to sacrifice and/or sample collection, and sample collection tubes (serum, plasma with anticoagulant, etc.).
Species Selection for PK Evaluation
Species used in PK testing must be suitable for in vitro/in vivo predictions and be clinically relevant. In drug discovery, selections are made based on the in vivo PK efficacy disease models used, relevant toxicology species, and species desired for allometric scaling. Preclinical toxicology testing is required in one rodent and one non-rodent species. Species typically used include mice, rats, rabbits, and guinea pigs for rodents, and dogs, minipigs, and non-human primates for non-rodents. Typically, the strain selected should be relevant to the in vivo PK efficacy models during early discovery, and should match an organization’s historical toxicological data for toxicology selections. Samples may be collected from preclinical pharmacology studies for drug PK analysis comparisons in the disease model if the model allows. Toxicology and TK assessments are typically tested in males and females to determine any gender differences, and usually include TK sample collections on the first and last days of the toxicology study to compare single to multiple dose exposures in the PK assays. The number of animals used in a study should be minimized for ethical considerations, but allow for the collection of sufficient information and statistics to meet the study objectives. PK PD in drug development plays a crucial role in optimizing dosing strategies and understanding drug behavior in the body. As with all in vivo protocols, study protocols are reviewed and approved by an Institute Animal Care and Use Committee (IACUC) to ensure the animal studies are performed safely and humanely.
Dose Administration Routes and Regimens
Routes of administration for dosing include intravenous bolus (IV), intravenous infusion, oral (PO), intra-peritoneal (IP), intramuscular (IM), inhaled or intratracheal (IT), or subcutaneous (SC or SQ). Typically, the route chosen for dose administration in PK and efficacy studies is chosen to match the desired route of delivery in the clinic. During early drug discovery this is not always the case – limitations in solubility or PK exposure may require differing dosing routes to achieve target proof-of-concept. For example, intraperitoneal dosing is often evaluated in early efficacy studies to demonstrate therapeutic activity if oral bioavailability is insufficient to reach efficacious exposures in order to demonstrate proof-of-concept of the compound class in the disease model. In these cases, PK drug testing will be performed by all potential routes prior to efficacy testing to determine the appropriate path forward. Successful demonstration of proof-of-concept will then trigger additional medicinal chemistry efforts to improve oral absorption while maintaining potency and selectivity, with PK assay evaluations on molecules of interest. Initial PK studies will typically include an intravenous dosing arm so that bioavailability may be established for the extravascular dosing routes. PK PD in drug development is essential for understanding the relationship between drug concentration and its pharmacological effects. Following lead identification, efficacy and PK should be demonstrated using the intended clinical route of dosing, and toxicology tests should also be performed using the intended clinical route of dosing, except in rare cases where dosing is target-specific and systemic exposures are negligible.
Dosing regimens are dependent on the intended objectives of the study. Most early studies are single dose to get an early assessment of a compound’s exposure and bioavailability and determine eligibility and regimens needed for efficacy model testing. Single dose PK in the efficacy species can be investigated and modeled to determine how often the compound requires dosing to maintain exposure or reach steady-state during chronic administration efficacy studies. PK PD in drug development is critical for understanding the relationship between dosing regimens and their therapeutic effects. TK studies often investigate single vs. multiple dose exposures. Keep in mind species differences when looking at PK and dosing regimens: if twice daily dosing is required for efficacy in a mouse model, it may not be necessary for clinical efficacy, as the average heart rate in a mouse ranges from 500-700 beats per minute, whereas in a human it is 60-100 beats per minute.
Dosing Vehicles
Formulations for administration are critical to a well-designed PK study. For IV administrations of a test article, a formulation must demonstrate solubility, as any solids in an IV formulation could result in unsafe cardiac or pulmonary embolisms. Solubility is desirable in early discovery for extravascular administrations as well since it makes data interpretation and compound comparisons more consistent. During later phases of drug discovery, various vehicles may be tested to best interpret the final product’s clinical exposure potentials. For example, if the intended product is an oral tablet, and early drug PK studies were dosed in solution, a suspension formulation should be tested to determine whether dissolution of the solid tablet may limit oral absorption and bioavailability. Vehicle excipients are typically selected to yield the desired solubility and safety. Novel excipients not previously tested are rarely used since they would require full toxicological evaluations before being used in a marketed product. During or prior to preclinical toxicology assessments, compound solubility, homogeneity, and stability should be demonstrated in all dosing vehicles ranging all tested concentrations.
PK Sample Collection
There are two main categories for PK sample collection used in protocols dependent upon the test model and study objectives, destructive and serial sampling. Destructive sampling uses a single animal sacrificed at each time point collected during the PK study. Destructive sampling is often used in mouse studies due to small total blood volume, or in studies where target organ tissue PK is being evaluated since serial tissue collection is typically unfeasible to obtain adequate biological samples for PK assays. Serial sampling involves multiple samples drawn from single animal for a complete time course after dosing. Serial sampling requires fewer test subjects and doesn’t require animal sacrifice at the end of study, so it is often used in the larger non-rodent species. Serial sampling also allows for cross-over designs, where the same subjects may be used to collect biological samples from different dose levels or dosing routes following a washout period. Blood is drawn via cannula, or direct bleed (retro-orbital, sub-mandibular, tail snip) for serial sampling. Terminal samples in rodents are typically collected by cardiac puncture under suitable anesthesia for destructive sampling or terminal serial sampling. Blood is drawn into serum collection tubes or tubes with anticoagulant (e.g. heparin, EDTA), maintained on ice, and centrifuged to obtain plasma, which may be stored frozen until processing and analysis of the drug PK assay.
Pharmacokinetics Evaluations
PK study assessment involves three major components: the in vivo PK study (dosing and sample collection, described above), sample bioanalysis for test article concentrations, and PK parameter calculations.
Sample Bioanalysis
Test article concentrations are typically measured in serum or plasma. Serum and plasma offer advantages over whole blood including ease of storage and handling, no cell lysis or clotting concerns, and the samples can be frozen/thawed with minimal impact. Stability of the test article must be demonstrated in any tested biological sample matrices for PK assays to demonstrate reliable PK predictions. Understanding pharmacokinetics vs pharmacodynamics examples in these matrices can help illustrate how drug concentrations and their effects are monitored and interpreted in various clinical and preclinical settings.
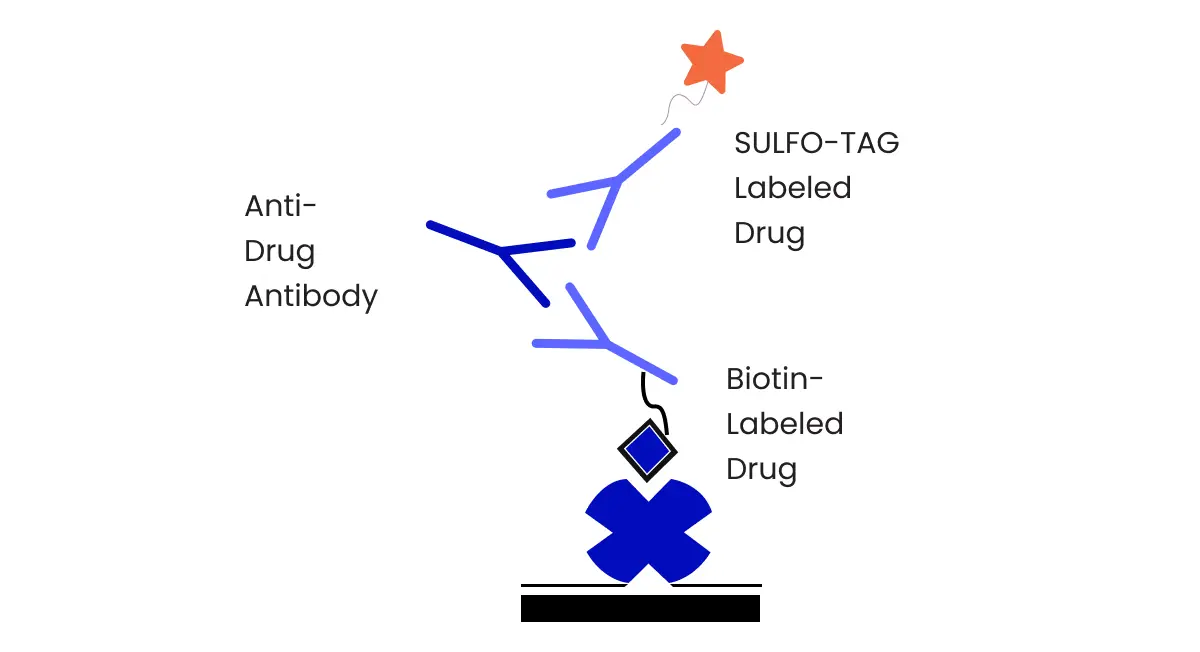
Analytical instrumentation used varies depending on the test article. Biological samples are not suitable for direct analysis on most analytical equipment used for analysis of pk assays. The chosen methodology must be suitable for the test article, matrix, and concentration range needed, and methods must be robust, accurate, precise, and reproducible. Analysis involves compound extraction, separation, or isolation and detection. Extraction may be performed by protein precipitation, liquid-liquid extraction, solid-phase extraction, or other techniques to recover the drug from the biological matrix and make the sample suitable for analysis on bioanalytical equipment. The majority of small molecules are analyzed via liquid chromatography for separation (HPLC, UPLC) with detection by mass spectroscopy (MS or MS/MS). Other detectors may be used and have been used historically, including UV/Vis, fluorescence, phosphorescence, liquid scintillation counters or other radio-detectors as appropriate, etc. Separation techniques may also employ various chromatographic separation methods, for example gas chromatography and thin-layer chromatography. The FDA’s bioanalytical guidance refers to these as chromatographic assays. For large molecules and biological therapeutics, ligand binding assays are typically used, including ELISA, RIA, MSD, etc. In these cases, compound isolation is achieved via binding to specific antibodies, resulting in a measurable signal change detected through an appropriate detector. Sample bioanalysis should include appropriate controls to ensure accuracy, precision, and stability. Observing pharmacokinetics vs pharmacodynamics examples during the analysis can help in understanding the practical implications of the methodology on drug behavior and effects. Additionally, comparing pharmacokinetics vs pharmacodynamics examples across different test articles can provide valuable insights into their analytical requirements and performance. Method validations in all matrices are required for preclinical toxicokinetics evaluations based on the FDA guidance for industry.
Pharmacokinetic Parameter and Statistics Calculations
Once sample concentrations have been determined, PD and PK analysis may be performed using the bioanalytical results from the PK assay. Drug concentrations vs. time are plotted, and pharmacokinetic parameters are calculated from the PK assay results. PK software packages such as Phoenix WinNonlin are used for efficient and reliable PK predictions. Typical non-compartmental PK parameters are defined in Table 1.
Table 1. Select Non-compartmental PK Parameters
| Parameter |
Description of Parameter |
| Tmax |
The time after dosing at which the maximum observed concentration of test article was observed |
| Cmax |
The maximum observed concentration of test article measured after dosing |
|
Cmax,n |
The dose-normalized maximum observed concentration of test article measured after dosing, calculated as Cmax/Dose |
| C0 |
The theoretical concentration at time 0 for an initial IV bolus dose extrapolated from initial measured plasma concentrations |
| C0,n |
The dose-normalized theoretical concentration at time 0 for an initial IV bolus dose calculated at C0/Dose |
|
AUC0-t |
The area under the concentration versus time curve from the start of dose administration to the time after dosing at which the last quantifiable concentration is observed, using the linear trapezoidal method |
|
AUCn,0-t |
The dose-normalized area under the concentration versus time curve from the start of dose administration to the time after dosing at which the last quantifiable concentration is observed, calculated as AUC0-t/Dose |
|
AUC0-∞ |
The area under the concentration versus time curve from time zero extrapolated to infinity (if data permits) |
|
AUCn,0-∞0 |
The dose-normalized area under the concentration versus time curve from time zero extrapolated to infinity, calculated as AUC0-∞/Dose |
| λz |
The terminal elimination or disposition rate constant |
|
t½ |
The terminal elimination or disposition half-life |
| CL |
The clearance rate of the analyte from the analyzed matrix (IV only) |
| Vss |
The volume of distribution of the analyte in the test system estimated at steady state (IV only) |
| MRT |
Mean residence time, a model-independent representation of the average time a compound resides in the body |
| F% |
Bioavailability, represents the fraction of a dose reaching systemic circulation intact; i.e. fraction of dose absorbed |
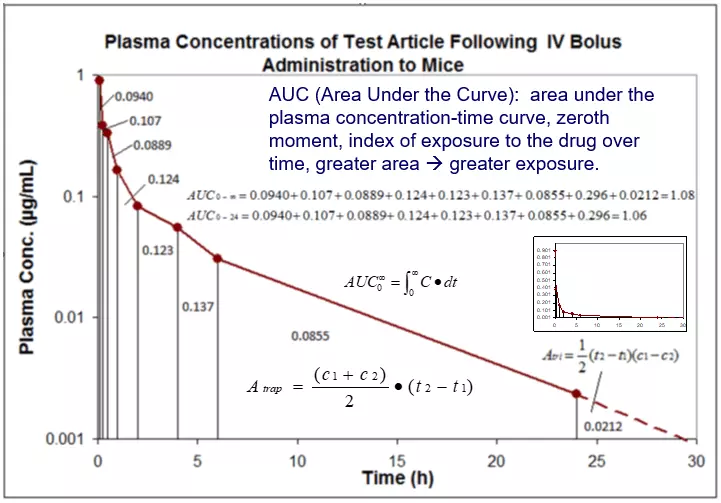
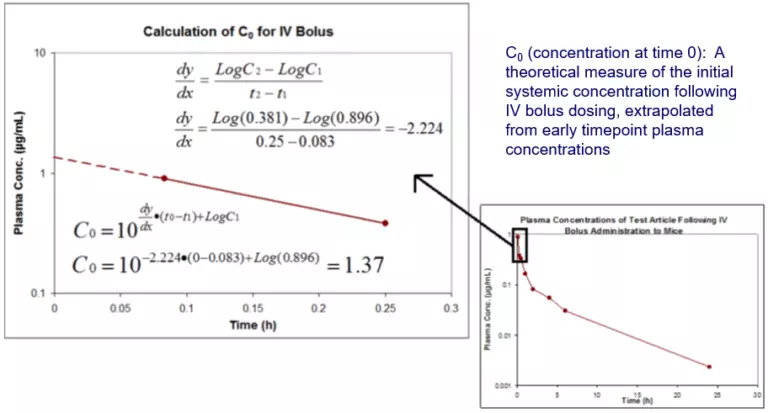
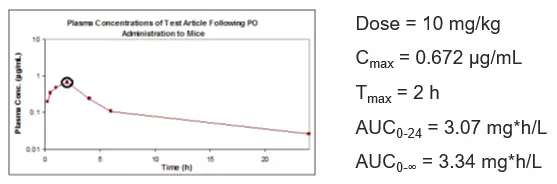
The most important PK parameters referenced when discussing exposure and activity or toxicity are PK AUC and pharmacokinetics Cmax or C0 for an IV dose. The area under the curve (AUC) represents the total exposure of the test article over the tested time course and can be extrapolated to infinity for well-designed studies. Figure 1 demonstrates how AUCs are calculated following an IV bolus dose. Following an IV bolus dose, one can assume the maximum systemic exposure occurs at the time of dosing; this concentration is referred to as C0. Figure 2 demonstrates extrapolation and calculation of C0 following an IV dose.
Figure 1. Calculation of PK AUC using the linear-linear trapezoidal rule with time extrapolation to infinity.
Figure 2. Extrapolation and calculation of C0 following an IV dose.
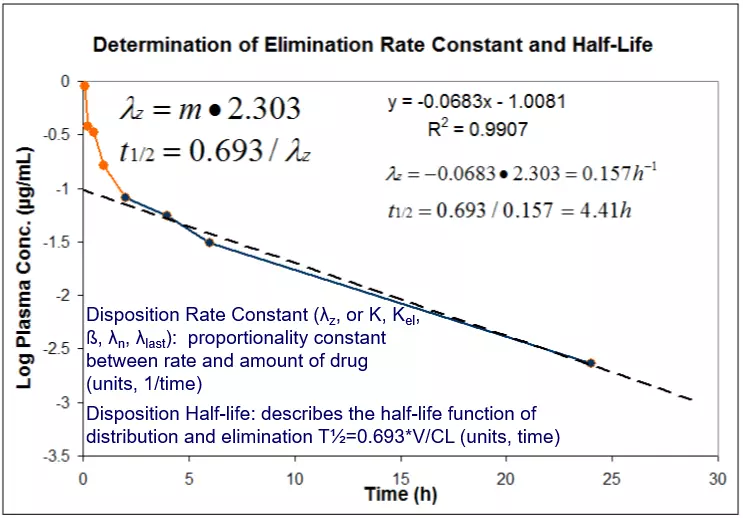
Certain PK parameters including clearance and volume of drug distribution can only be calculated following an IV dose. Similar parameters (apparent clearance CL/F, apparent volume V/F) may be calculated for oral or other extravascular dosing routes, but contain a correction factor for bioavailability. While elimination half-life may be calculated for any dose route, it is best represented for a compound following an IV dose since with direct systemic administration the absorption component is bypassed. The elimination rate constant and half-life are determined as demonstrated in Figure 3.
Figure 3. Calculation of Elimination Rate Constant and Half-Life.
Systemic Clearance (CL or CLs) represents the rate at which a drug is removed from systemic circulation. Measured clearance values are determined from IV dosing, and represent total additive clearance from all potential clearing organs (e.g. liver, kidneys, etc.). Clearance values are measured in units of volume/time, so they can be thought of as a proportionality constant between plasma concentration and the rate of elimination. For the data represented in the figures above, the calculation is performed as follows:
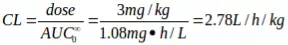
Apparent Volume of Distribution (Vd, Vz, Vß or Varea): The apparent volume of distribution reflects the reversible uptake of drug distribution by tissues from the blood. It can be thought of as a fictitious volume that a drug occupies in the body as a uniform sack relative to the drug concentration in blood. Large volumes of distribution indicate greater concentrations of the drug reaching tissues relative to measured plasma concentrations, and can indicate the compound’s potential for reaching the desired target. For the data represented in the figures above, the calculation is performed as follows:
The majority of PK parameters can be represented mathematically to statistical moment (or residence time) analyses of the first two moment curves of the time vs. concentration data.
PK Parameters derived from S.H.A.M. measures: Slope, Height, Area, and Moment
| Slope: |
λz and t½ |
| Height: |
V |
| Area: |
CL (with dose)
Vz (with dose, to get CL, and λz)
Vss (with dose, to get CL, and MRT)
F (with another area, extravascular) |
| Moment: |
MRT (mean residence time, with area)
Vss (with CL and MRT) |
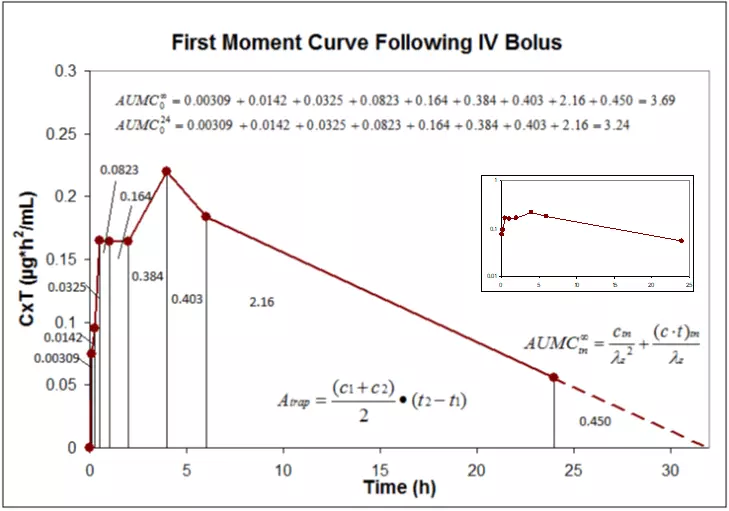
Calculation of moments and residence times requires calculation of AUCs under several curves (typically zeroth and first moments done for noncompartmental PK analysis for PK AUC and AUMC, respectively). The first moment curve is generating by plotting the independent variable (time) by the factor of the dependent and independent variables (concentration x time).
Another PK parameter calculated but not often reported is the area under the moment curve (AUMC). While this parameter is not useful on its own, it can be used to calculate other meaningful PK parameters such as volume of distribution at steady-state (Vss) and mean residence time (MRT). Figure 4 demonstrates the calculation of AUMC for the IV data shown in Figures 1-3.
Figure 4. Calculation of AUMC using the first moment curve.
PK Parameters derived from first moment analysis:
Mean Absorption Time (MAT, also referred to as mean input time): The average amount of time a compound stays at the absorption site (0 for IV bolus, ½ time in syringe for IV infusion); estimates rate of absorption independent of model. For a first-order input process, MAT = 1/Ka where Ka is the 1st order absorption rate constant. For an extravascular dose, MAT is the difference in MRT for the dose in question relative to that for an IV bolus.
Mean Residence Time (MRT): A model-independent representation of the average time a compound resides in the body. MRT is another measure of elimination and is an example of a statistical moment. For a simple one-compartment model with 1st order disposition, MRT = 1/K where K is the 1st order elimination rate constant.
Apparent Volume of Distribution at Steady State (Vss): The apparent volume of distribution reflects the reversible uptake of drug by tissues from the blood. At steady state, this is an estimate of drug distribution independent of elimination processes. It is most useful for predicting the plasma concentrations following multiple dosing to a steady-state where rate in = rate out. Vss is proportional to the amount of drug in the body versus the plasma concentration of the drug at steady state.
PK Parameters following extravascular doses:
Following an oral or extravascular dose, similar techniques as those described above are used to determine AUCs and AUMCs:
Area under the Curve and Maximum Concentration (AUC and Cmax): PK AUC is calculated for an oral dose in the same fashion as for an IV dose. The pharmacokinetics Cmax is the highest measured concentration, and the time at which it is achieved is referred to as Tmax. Other extravascular routes are measured the same way; the key difference is that an IV bolus delivers the drug directly into systemic circulation, bypassing any absorption mechanisms, thereby giving a reference point for maximum bioavailability.
Absolute Bioavailability (F): The fraction of a dose reaching systemic circulation intact; i.e. fraction of dose absorbed (not to be confused with relative bioavailability, which is the fraction of a dose of drug reaching systemic circulation relative to a reference product or alternative dosing regimen/vehicle (e.g. relative bioavailability of a suspension to solution dose).
Pharmacokinetics results interpretation
In summary, the parameters described above can be used to describe the PK process pharmacokinetics vs pharmacodynamics ADME in the test subjects. PK prediction is typically performed using PK software packages such as Phoenix WinNonlin, or packages in a LIMS or other data management systems designed for PK/TK analysis. Absorption is represented by AUC, pharmacokinetics Cmax, Tmax, and bioavailability. Drug distribution is represented by volumes of distribution. Metabolism and elimination are represented by clearance and elimination half-life. AUCs, mean residence times and half-lives can be used to estimate dosing regimens for efficacy and scale for estimates of toxicity. PK data can be directly compared to measurable efficacy endpoints to establish an understanding of a compound’s PD and PK relationship. Generally speaking, there is no such thing as good or bad PK results – interpretation depends upon the therapeutic area, drug target affinity, drug target location, and toxicity or establishment of therapeutic index. For most non-systemic drug targets, low clearance, moderate PK AUC and Cmax values, and high volumes of drug distribution are desirable, but for a bloodborne disease such as leukemia, low volumes of distribution may be more desirable. For cytotoxic chemotherapeutic agents targeting solid tumors, high clearance and high distribution volumes may be desirable. PK drug scientists in early discovery are often asked if efficacy is “Cmax or AUC driven” following early preclinical pharmacology assessments – this question is less about PK and relates more to the test article’s affinity and reversibility with the molecular disease target. Desirable PK should normally include adequate systemic exposure required for target engagement and efficacy, a wide therapeutic index between minimum efficacy and tolerability, and adequate exposure for maintaining therapy on a reasonable readily compliant dosing regimen in the clinic.